
BRAF靶點抑制劑中國臨床研究現(xiàn)狀
1.靶點機制
BRAF 基因位于染色體 7q34,編碼絲氨酸/蘇氨酸蛋白激酶,是 RAF 家族成員。BRAF蛋白與 KRAS 蛋白同為 RAS-RAF-MEK-ERK 信號通路中上游調(diào)節(jié)因子,使 MEK 蛋白磷酸化,隨后 ERK 蛋白磷酸化,系激活參與細胞增殖和生存的相關(guān)基因。突變的 BRAF 蛋白增強了激酶的活性,可在體外轉(zhuǎn)化。而其中具有致癌以及治療價值的是 V600 突變,主要包括 V600E 和 V600K 突變。該位點的突變可引起下游活化致癌,占整體 BRAF 突變的一半。大多數(shù) BRAF 突變的患者既往有吸煙史,病理類型是腺癌。BRAF 突變一般與 EGFR、KRAS 等突變相互獨立和排斥,并不同時出現(xiàn)。
按照作用靶點的不同,BRAF 抑制劑分為多靶點激酶抑制劑和 BRAF V600E(單靶點)抑制劑兩類。多靶點激酶抑制劑,如索拉非尼(Sorafenib) 、瑞戈非尼(Regorafenib) 、培唑帕尼(Pazopanib)、ASN-003 和 CEP-32496 等(詳見多靶點 TKI 部分),對于包括 BRAF 在內(nèi)的多種激酶均有抑制作用,具有廣譜的抗腫瘤及抗血管生成作用,屬于非特異性 BRAF抑制劑。特異性 BRAF V600E(單靶點)抑制劑,如維莫非尼(Vemurafenib)、達拉菲尼(Dabrafenib)、PLX-8394 和康奈非尼(Encorafenib)等,對 BRAF 尤其是 BRAF V600E 有很高的抑制活性,目前主要獲批用于治療黑色素瘤。
2.中國臨床研究申報現(xiàn)狀
目前國內(nèi)進行中的 BRAF 抑制劑臨床研究共 20 余項。其中Ⅱ期臨床研究有 4 項,比如達拉非尼聯(lián)合曲美替尼治療 BRAF 突變陽性肺癌的研究,DCC-2618 在胃腸道間質(zhì)瘤患者中的有效性、安全性及 PK 特征的研究, Encorafenib 聯(lián)合西妥昔單抗治療 BRAF 突變晚期腸癌的研究等。目前Ⅰ期臨床研究共 5 項,如 RX208 在晚期惡性實體腫瘤患者中的Ⅰ期臨床研究,TQ-B3233 膠囊Ⅰ期耐受性和藥代動力學臨床試驗,評價單次和重復口服給藥的藥代動力學和安全性,還有評價 GSK2118436 單藥和聯(lián)合給藥的藥代動力學和安全性等。
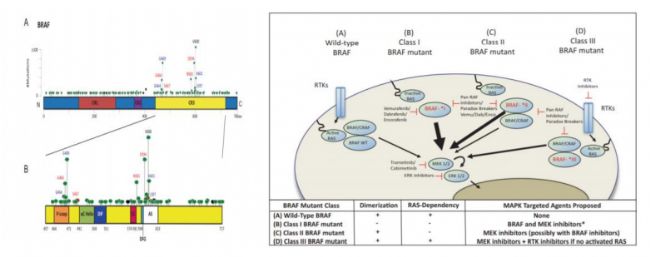
資料來源:DANKNER M, ROSE AAN, RAJKUMAR S,et al. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations[J]. Oncogene,2018,37(24):3183-3199. doi:10.1038/s41388-018-0171-x. Epub 2018 Mar 15. PMID: 29540830.
3.簡評
BRAF 蛋白是 MAPK/ERK 信號通路中重要的上游調(diào)節(jié)因子,其 V600E 突變可激活下游 MEK 蛋白,進一步引起腫瘤細胞生長、增殖和侵襲,易發(fā)生于結(jié)直腸癌、甲狀腺癌、黑色素瘤等多個癌種之中,該類患者往往預后較差。雖然第一代 BRAF 抑制劑(Vemurafenib 和 Dabrafenib)在 BRAF V600E 突變的黑色素瘤患者上取得了良好的效果,但是用于其他腫瘤患者的 BRAF V600E 突變(如結(jié)直腸腫瘤)效果卻不佳,且在一年之內(nèi)均產(chǎn)生抗藥性。因此,BRAF V600E 抑制劑往往要聯(lián)合 EGFR 單抗或 MEK 抑制劑,才能夠有效改善該類患者的生存現(xiàn)狀及預后(如 Encorafenib 與西妥昔單抗或 Binimetinib 組合療法)。新一代的 BRAF 抑制劑(如 PLX-8394、CEP-32496 等),也成為現(xiàn)階段的研發(fā)方向之一。BRAF 作為抗腫瘤治療中一個熱門的分子靶點,盡管目前已有 20 多項臨床研究正在進行中,但是其中Ⅱ、Ⅲ期研究仍然較少,高效的靶點抑制劑值得進一步探索。



Copyright(C) 1998-2024 生物器材網(wǎng) 電話:021-64166852;13621656896 E-mail:info@bio-equip.com