
定量蛋白質組學質譜采集技術進展
張偉*
(賽默飛世爾科技(中國)有限公司, 上海201206)
摘要 質譜是定量蛋白組學的主要工具。近年來隨著定量蛋白質組學研究的深入,傳統(tǒng)質譜定量技術面臨著復雜基質干擾、分析通量限制等諸多問題。而最近一系列質譜新技術的發(fā)展,包括同步母離子選擇(SPS)、質量虧損標記、平行反應監(jiān)測(PRM)、多重累積(MSX)和多種全新數據非依賴性采集(DIA)等,為解決目前蛋白質組學在相對定量和絕對定量分析方面的局限提供了有效途徑。本文對定量蛋白質組學目前遇到的瓶頸問題進行了分析,總結了質譜定量采集技術的最新進展,并評述了這些新技術的特點以及在定量蛋白質組學應用中的優(yōu)勢。
關鍵詞 定量蛋白質組學; 同步母離子選擇; 平行反應監(jiān)測; 數據非依賴性采集; 綜述
1引言
當今蛋白質組學的關注焦點和研究趨勢已經逐漸從定性分析轉向定量分析。定量蛋白質組學是對細胞、組織乃至完整生物體的蛋白質表達進行定量分析,對生物過程機理的探索和臨床診斷標志物的發(fā)現與驗證具有重要意義[1,2] 。定量蛋白質組學分為相對定量與絕對定量[3] 。相對定量即差異比較,通過質譜大規(guī)模、高通量地對兩種或多種不同生理、病理條件下的樣本進行定量分析,獲得蛋白質表達量的精確差異,主要方法有穩(wěn)定同位素標記和非標記兩種技術手段[4,5] 。絕對定量即獲得蛋白的具體表達量,利用質譜監(jiān)測目標蛋白的專一性肽段(Unique Peptide)獲得色譜-質譜峰面積,并與已知量的標準肽段(外標法)或穩(wěn)定同位素標記的重標肽段(內標法)比較確定具體量,實現絕對定量。主要質譜方法是對專一性肽段進行選擇反應監(jiān)測或稱多反應監(jiān)測(Selected/ Multiple reaction monitoring, SRM/ MRM)[6] 。
穩(wěn)定同位素標記技術是蛋白質組學相對定量的經典方法。樣本在穩(wěn)定同位素標記后、質譜分析前混合,一次分析實現差異定量,有效消除了色譜和質譜分離分析過程中的不穩(wěn)定性,最大程度減小了定量誤差。常見方法有基于代謝標記的SILAC[7] 、基于酶解標記的18 O 標記[8] 和基于化學標記的二甲基化[9] 等,這些方法通過一級母離子提取峰面積實現定量比較。但是,一級定量具有標記通量低、動態(tài)范圍差、靈敏度不高等不足,因此, 近年來,基于同重同位素標記的二級定量方法使用越來越廣泛[10] 。利用同重同位素標簽標記肽段,在一級質譜不同樣本的肽段分子量沒有區(qū)分,相互疊加,提高了靈敏度;二級碎裂獲得分子量不同的報告離子,在b/ y 離子定性的同時,通過報告離子之間的強度差異實現定量,提高了動態(tài)范圍。同重同位素主要標記試劑有iTRAQ[11] 和TMT[12] ,標簽容量分別達到了8 標和6 標。然而,同重同位素標記技術面臨共洗脫肽段干擾的問題。蛋白質組學樣本非常復雜,在色譜上存在大量共洗脫肽段,而質譜在選擇母離子進行二級分析時,選擇窗口通常在m / z 2 左右,分子量接近的共洗脫肽段被同時選擇,碎裂出的報告離子與目標肽段報告離子疊加,降低了定量比例的準確性[13,14] 。Ting 等[15] 研究證明,在復雜樣本中,共洗脫肽段嚴重干擾了報告離子的強度,造成肽段和蛋白的定量比例低于真實比例,產生“低估效應冶。這一問題已成為同重同位素標記定量技術的瓶頸。
基于三重四極桿的SRM(或稱MRM)是質譜定量的金標準,在蛋白質絕對定量中也廣泛使用[6] 。SRM 根據專一性肽段的母離子質量和子離子質量,第一級質量分析器(Q1)篩選母離子,進入碰撞池碎裂后,第二級質量分析器(Q3)再篩選子離子,最大程度地去除干擾離子,監(jiān)測母離子-子離子形成的離子對的信號響應。通過外標法,利用已知量的標準肽段繪制標準曲線; 或內標法,直接加入已知量的同位素重標肽段同時監(jiān)測,從而實現定性確證和定量檢測[6,16] 。SRM 靈敏度高、線性范圍廣,是目標蛋白驗證和絕對定量的有效手段。然而,隨著定量蛋白質組學的深入發(fā)展,樣本基質越來越復雜、目標蛋白豐度越來越低,容易受到高豐度蛋白的掩蓋。而SRM 由于質量分辨率低,難以有效去除復雜基質背景的干擾,易造成假陽性[17,18] 。另一方面,隨著分析通量的要求越來越高,一次分析可能需要監(jiān)測成千上萬個離子對,而SRM 速度和靈敏度的局限使得能同時監(jiān)測的離子對數量有限[19] ; 此外,離子對、碰撞能量等條件的優(yōu)化也費時費力,難以滿足目標蛋白質組學高通量發(fā)展的需要,特別是大樣本量的生物標志物和系統(tǒng)生物學研究[20,21] 。因此,蛋白質絕對定量同樣面臨著較大的技術挑戰(zhàn)。
近兩年來,隨著以Orbitrap 為代表的高分辨質譜硬件技術不斷進步、采集方法不斷創(chuàng)新,定量蛋白質組學遇到的諸多瓶頸正逐步得到解決。這些技術包括基于同重同位素標記技術的同步母離子選擇和質量虧損標記,相對于傳統(tǒng)SRM 掃描的高分辨平行反應監(jiān)測和多重累積-平行反應監(jiān)測,以及多種全新數據非依賴性采集技術。
2同重同位素標記進展:同步母離子選擇與質量虧損標記技術
以iTRAQ 和TMT 為代表的同重同位素標記技術面臨著兩方面的技術瓶頸:(1) 大量共洗脫肽段對定量準確性的影響:分子量接近的共洗脫肽段與目標肽段共同選擇并碎裂,報告離子疊加,造成實際定量比例不能準確反映真實結果[13 ~15] ; (2) 等重同位素標簽通量有限:以iTRAQ 為例,其報告基團(N-methylpiperazine)由6 個C 和2 個N 組成,因此容量上限為8 標,無法完成更高通量樣本的分析[22] 。而通過增大報告基團來提升標簽容量又會造成靈敏度的下降[23] 。
為解決共洗脫肽段干擾的問題,Ting 等[15] 使用LTQ-Orbitrap 進行MS3 掃描,對TMT 標記的樣本進行分析(圖1)。其中MS2 使用能量較低的CID (35%)碎裂,獲得b/ y 離子碎片用于序列鑒定,同時確保了TMT 標簽不會過多碎裂。在母離子m / z 110 ~160% 的范圍內,選擇最強的一個碎片離子,進行高能量HCD (60%)碎裂和MS3 檢測,使報告基團充分碎裂,獲得報告離子比例信息。結果表明,通過母離子和子離子雙重篩選,共洗脫肽段的干擾能夠被徹底去除,獲得的定量比例與理論比例一致。例如,在摻入等比例人源蛋白的不同比例酵母全蛋白樣本中,理論比值為10頤1,傳統(tǒng)MS2 測得的比值僅約為3,而MS3 測的比值為11. 7,與理論比值非常接近。
但是與傳統(tǒng)MS2 方法相比,MS3 信號強度明顯降低。因此,雖然MS3 的定量準確度大大提高,但定量靈敏度卻明顯下降,也使得MS3 定量方法并沒有普遍應用到實際分析中[24] 。此外,近來發(fā)展的TMT互補離子(TMTc) [25] 和QuantMode[26] 等技術雖然也能降低共洗脫肽段的干擾,但過程較為繁瑣,準確度無法與MS3 方法相媲美。
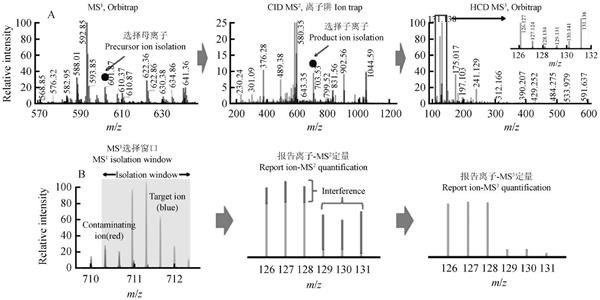
圖1MS3 定量方法示意圖[15] :(A) MS3 方法質譜采集過程; (B) MS3 與傳統(tǒng)MS2 方法結果對比
Fig. 1Schematic elucidation of MS3 method[15] : (A) Acquisition workflow of MS3 ; (B) Comparison of MS3 and classic MS2 results.
同步母離子選擇(Synchronous precursor selection, SPS)技術的出現徹底解決了MS3 響應弱的問題。SPS 技術利用多頻切跡的選擇波形電壓(MultiNotch) [27] ,在線性離子阱中實現一次選擇同時獲得多個離子,最多可同時選擇15 個(圖2A)。利用這一原理,SPS 技術在MS2 中同時選擇目標肽段的多個b/ y碎片進行MS3 分析,產生的報告離子相累積,使MS3 信號響應大幅提高(圖2B)[24,28] 。實驗結果表明,使用SPS 進行MS3 定量分析,即二級CID 碎裂,并同時選擇多個碎片離子進行三級HCD 碎裂和Orbi-trap 檢測,獲得的MS3 譜圖響應與傳統(tǒng)MS2 譜圖相當,定量成功率超過70%,與傳統(tǒng)MS2 定量結果相當(圖2C)[24,28] 。同時,多個碎片報告基團的疊加相比傳統(tǒng)MS3 單個碎片報告基團,得到的比值更加穩(wěn)定,定量重現性和準確度進一步增加(圖2D)。此外,相比傳統(tǒng)方法,MS3 定量方法只能用于Lys-C 酶解樣品,SPS MS3 定量方法適用于廣泛使用的Trypsin 酶解樣品,能更普遍用于常規(guī)定量蛋白質組學[29] 。Weekes 等[30] 使用SPS MS3 技術定量研究了巨細胞病毒(CMV)感染纖維母細胞的過程和機理,獲得超過8000 個蛋白(包括1184 個細胞表面蛋白)在8 個時間點感染過程中的精確定量變化,實現了實時動態(tài)定量分析,開創(chuàng)了定量時序病毒組學(Quantitative Temporal Viromics)領域的先河。
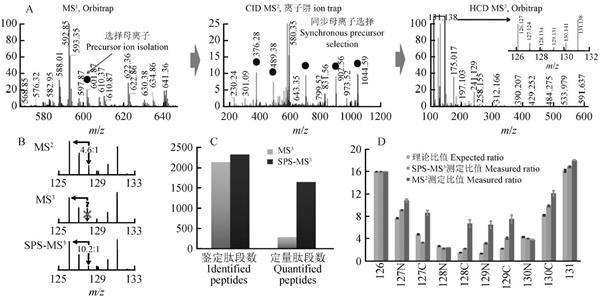
圖2 SPS MS3 定量方法示意圖[24,28] :(A) SPS MS3 方法質譜采集過程; (B) 傳統(tǒng)MS2 、MS3 、SPS MS3 對報告離子的影響; (C) MS3 、SPS MS3 定量靈敏度對比; (D) 傳統(tǒng)MS2 、SPS MS3 定量準確度對比
Fig.2 Schematic elucidation of synchronous precursor selection (SPS) MS3 method [24, 28] :(A) Acquisition workflow of SPS MS3 ; (B) Influence of classic MS2 ,MS3and SPS MS3 for reporter ion intensity; (C) Comparison of sensitivity of MS3 and SPS MS3 ; (D) Comparison of accuracy of classic MS2 and SPS MS3
對于同重同位素標簽通量的限制,Dephoure 等[31] 嘗試使用3 標SILAC 結合6 標TMT,同時定量18 組平行樣本,取得了良好的效果。而近年來,質量虧損標記技術的運用,從根本上拓展了報告基團的標簽容量。13C 和12C、15N 和14N 之間因核結合能形成質量虧損,巧妙利用13C14 N 與12C15N 之間6. 32 mDa的微小質量差,通過13C 與15N 相互替換,即可實現標簽容量上限的突破(圖3A)。例如,將TMT 6 標中TMT127、TMT129 報告基團上一個13 C 替換成15 N,形成TMT127L (12 C8 H16 15N1+ , 127. 1247608 Da) /TMT127H (12C713C1H1614N1+ , 127. 1310808 Da), TMT129L (12C613C2H1615N1+ , 129. 1314705 Da) /TMT129H (12C5 13C3H16 14N1+ , 129. 1377904 Da),標簽容量提升至8 標[32] 。類似地,在不改變報告基團結構的情況下,TMT 10 標、TMT 18 標均得以實現,有效解決了多組平行樣本的定量分析問題[33, 34] 。但是另一方面,6.32 mDa 的質量差異需要質譜在m / z 100 ~200 范圍達到50000 以上的分辨率才能實現基線分辨(圖3B),普通高分辨質譜難以勝任[33] 。而基于傅里葉變換原理的超高分辨Orbitrap 和FT-ICR,分辨率與m / z 成反比,能夠輕松分辨m / z 100 ~200 的質量虧損標簽[34] 。目前,基于質量虧損標記的TMT 10 標已經商品化,廣泛應用于系統(tǒng)生物學和臨床標志物研究等大樣本量的定量蛋白質組學(圖3C)。

圖3質量虧損標記提升標簽通量[33] :(A) 質量虧損標記原理(TMT-10); (B) 區(qū)分6 mDa 所需的質量分辨
率; (C) 質量虧損報告離子質譜示意圖(TMT-10)
Fig. 3Increasing labeling capacity by mass defect isotopes [33] :(A) Principle of mass defect labeling (TMT-10);(B) Minimum resolution for discrimination of 6 mDa mass difference; (C) Mass spectrum of mass defect reporter ions(TMT-10)
近年來,以同步母離子選擇SPS 和質量虧損標記為代表的新技術的發(fā)展,使得傳統(tǒng)同重同位素標記定量面臨的諸多問題正逐步獲得解決,成為蛋白質組學相對定量的有力工具和發(fā)展趨勢。
3目標蛋白定量進展:平行反應監(jiān)測與多重累積
SRM 使用低分辨四極桿進行兩級離子篩選和監(jiān)測,在小分子分析中能有效區(qū)分目標化合物和基質背景,因為小分子的化學結構通常差異較大[35] 。然而在蛋白質組學中,由于肽段性質的相似性,SRM 難以完全排除復雜樣本的背景離子,容易受到較大干擾,影響蛋白絕對定量的準確性和專一性[17,18] 。此外,SRM 還需要花費相當的精力進行離子對的選擇和優(yōu)化[36] 。質譜技術的改進也為SRM 帶來改善。高分辨SRM (H-SRM)將四極桿的母離子選擇窗口從傳統(tǒng)的m / z 0. 7 縮小到m / z 0. 2, 從而有效地降低基質背景的干擾[37,38] 。然而,選擇窗口縮小也同時降低了選擇效率,因此該技術更多應用于小分子分析。此外,通過增加第三級質量篩選形成三級MRM (MRM3 )掃描,也有效提高了選擇性[39] 。但是MRM3 面臨著同樣的問題,額外一級離子篩選降低了檢測靈敏度,同時也增加了循環(huán)時間。
四極桿-高分辨串聯(lián)質譜技術的發(fā)展和掃描速度的提升,為目標蛋白定量提供了新的途徑。平行反應監(jiān)測(Parallel reaction monitoring, PRM)正是相對于傳統(tǒng)SRM 建立起來的高分辨離子監(jiān)測技術[40,41] 。PRM 基于以Q-Orbitrap 為代表四極桿-高分辨質譜平臺,與SRM 每次掃描一個Transition 不同,PRM 每次對一個母離子產生的所有Transition 進行全掃描,即平行監(jiān)測了一個母離子對應的所有離子對。首先,PRM 使用四極桿(Q1)選擇母離子,選擇窗口通常m / z 臆2; 然后,母離子在碰撞池(Q2)中碎裂,形成子離子; 最后,以Orbitrap 取代Q3,對所有子離子進行高分辨/ 高質量精度(HR/ AM)的全掃描,完成PRM 采集(圖4)[42] 。相比傳統(tǒng)的SRM,PRM 的優(yōu)勢在于:(1) 高分辨子離子監(jiān)測,質量精度達ppm 級(通常外標法<3 ppm,內標法<1 ppm),在不損失靈敏度的情況下,最大程度地排除了背景干擾; (2)二級為全掃描,無需事先確定離子對和優(yōu)化碰撞能量,只需要在數據處理時選擇響應最高的一個或幾個子離子,即可提取母子離子對的色譜峰進行定量,線性范圍可達5 ~ 6 個數量級; (3) 同時定性與定量分析,二級全掃描譜圖用于定性分析,選擇其中最佳的子離子提取離子對即可完成定量分析。
Peterson 等[40] 利用14 條標準肽段比較了SRM和PRM 兩種掃描方式的選擇性和靈敏度,結果表明在無基質的情況下,SRM 和PRM 的定量限均能達到amol 級; 而在酵母基質中,PRM 的定量限仍保持在amol 級,但SRM 的定量限只有fmol 級,上升了一個數量級(圖5A),SRM 離子對受到很大的基質干擾(圖5B)。Gallien 等[42] 使用SRM 和PRM 考察了35 條重標肽段的175 對離子對在尿液基質中的最低定量限,結果表明,只有19% 的離子對其SRM 定量限低于PRM 定量限。近來,PRM 逐漸應用到生命科學研究中,Tsuchiya 等[43] 對野生型和UFD4 基因(泛素融合降解通路)敲除型細胞中脯氨酸-茁-半乳糖苷酶(Ub-茁-bgal)的泛素化進行PRM 監(jiān)測和定量分析,研究兩種細胞中泛素鏈與蛋白連接位點的變化,證明了UFD4 與K29 型連接的泛素化密切相關。Tang 等[44] 利用PRM 驗證了組蛋白H3 和H4 的甲基化、乙�;⒈;�55 種修飾位點,并對修飾程度進行了定量分析,還發(fā)現了H3 K18 甲基化、H4 K20 乙�;戎T多全新修飾位點。
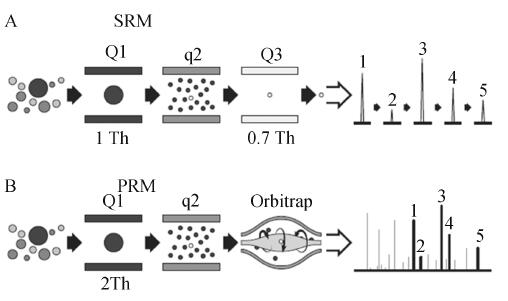
圖4選擇反應監(jiān)測SRM(A)與平行反應監(jiān)測PRM(B)原理[40]
Fig. 4Principle of selected reaction monitoring (SRM)
and parallel reaction monitoring (PRM) [40
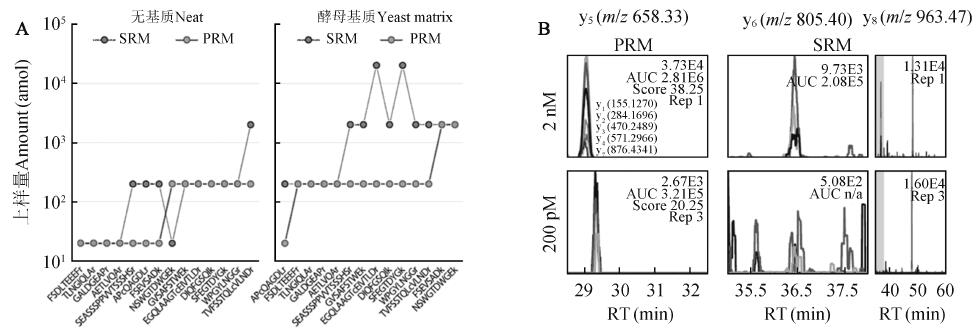
圖5SRM/ PRM 靈敏度與選擇性比較[40] :(A) 多種肽段在無基質和酵母基質中的定量限比較; (B) 酵母基質
中肽段GVSAFSTWEk 的提取離子流圖比較
Fig. 5Comparison of sensitivity and selectivity of SRM/ PRM [40] : (A) Comparison of LOQs of peptides in neat and yeast matrix. (B) Comparison of extracted ion chromatograms of peptide GVSAFSTWEk in yeast matrix
然而PRM 的分析通量不如SRM。PRM 能同時監(jiān)測最多10 ~15 個母離子,而SRM 能同時監(jiān)測上百個離子對,因為高分辨質譜的有效掃描速度通常只有10 ~15 Hz,遠慢于SRM 的有效掃描速度。但是這一問題正逐步得到解決,多重累積(Multiplexing, MSX)技術的發(fā)展和使用有效提高了PRM 的分析通量。多重累積利用Orbitrap 前端的C 型阱(C-trap),不同質量的母離子依次被四極桿選擇并儲存于C-trap 中,等待Orbitrap 完成上次掃描后,C-trap 中儲存的混合離子同時注入到Orbitrap 中進行掃描,實現最多10 個目標物的同時監(jiān)測,分析通量最高可提升10 倍[45] 。Gallien 等[46] 將多重累積與PRM 結合建立MSX-PRM 技術,利用4 重累積和保留時間分段(1. 5 ~2. 5 min 窗口),在60 min 液相梯度下,定量監(jiān)測了酵母全蛋白裂解液中770 個目標肽段,對應436 種蛋白。其中,同一保留時間下的肽段數量最高達60 個,在這種情況下,循環(huán)時間保持在2 s 以下,分析通量達到SRM 的水平。此外,通過優(yōu)化分辨率、離子注入時間等參數,目標肽段分析通量還能進一步提高:在MSX 設為8 時,同時監(jiān)測的肽段數量可達128 個[42,45] 。
綜上所述,高分辨PRM 和MSX-PRM 技術的出現,解決了傳統(tǒng)SRM 技術在定量蛋白質組學方面存在的諸多不足,為復雜樣本中目標蛋白的驗證和絕對定量提供了有效手段。
4數據非依賴性采集進展:基于Orbitrap 的多種全新DIA 技術
基于數據依賴性采集(Data dependent acquisition, DDA)的鳥槍法(Shotgun)是蛋白質組學的經典策略,也是蛋白質組學相對定量的主要技術手段。DDA 二級采集取決于一級掃描結果,易造成低豐度肽段的丟失,具有一定的隨機性,掃描點數也不均勻。而SRM 等目標采集方法不能采集非目標肽段,而且分析通量有限。近年來發(fā)展的數據非依賴性采集(Data independent acquisition, DIA)結合了DDA 與SRM 的特點,將整個掃描范圍等分為若干窗口,每個窗口依次選擇、碎裂,采集窗口內所有母離子的全部子離子信息。DIA 無需指定目標肽段,通量無上限,掃描點數均勻,利用譜圖庫即可實現定性確證和定量離子篩選,同時數據可以回溯,相比傳統(tǒng)DDA 和SRM 具有明顯優(yōu)勢[47] 。
Venable 等[48] 使用線性離子阱(LTQ),以10 m / z 窗口步長依次選擇、碎裂、采集,對15 N 標記的酵母全蛋白樣品進行相對定量分析,開創(chuàng)了DIA 應用的先河。結果表明,通過DIA 提取的色譜峰,其信噪比、重現性和定量準確性均明顯優(yōu)于DDA,蛋白定量數量增加了87%。Gillet 等[49] 基于Q-TOF (Trip-leTOF),采用32 個連續(xù)的m / z 25 窗口建立SWATH 技術,并證明SWATH 的選擇性與SRM 相當。SWATH 的發(fā)展使DIA 越來越多地應用于定量蛋白質組學[50 ~52] 。同時,基于Q-Orbitrap (Q Exactive)的DIA 技術,同樣使用25 m / z 步長,通過更高的分辨率,使復雜樣本定量的選擇性進一步提高。此外,諸如MSE、AIF 等質量范圍不分段的DIA 方法在蛋白定量中也有所應用,但由于這些方法不進行質量分段選擇,而是將所有離子同時碎裂、檢測,基質干擾較為嚴重,難以勝任復雜樣本的定量分析[49,53] 。
然而,由于掃描速度的限制,基于高分辨質譜的DIA 通常需要以m / z 25 的大窗口步長分段,以確保獲得足夠的掃描點數進行定量。而m / z 25 的選擇窗口仍然會引入較多的干擾離子,影響DIA 的定量分析效果(圖6A)[54] 。
近來,全新DIA 技術的發(fā)展使DIA 的選擇窗口不斷縮小,DIA 定量選擇性、靈敏度和重現性進一步提高。其中,前文提到的多重累積技術已應用到DIA 中,Egertson 等[54] 利用Q-Orbitrap 的多重累積功能發(fā)展了MSX-DIA 技術,隨機將5 個m / z 4 窗口依次選擇、累積,然后同時注入Orbitrap 進行掃描。MSX-DIA 總步長仍然是m / z 20,不影響掃描速度,而m / z 20 由5 段獨立的窗口組成,實際選擇窗口僅m / z 4(圖6A)。數據利用Skyline 軟件去卷積,即可將每個碎片歸屬到特定窗口中,實現m / z 4 窗口的選擇性(圖6B)。MSX-DIA 的選擇性已接近DDA,最大程度減少了共流出肽段和雜質的干擾。Q-OT-qIT 三合一質譜技術的出現為提高DIA 性能帶來了更多可能[55] 。利用Q-OT-qIT 中Orbitrap和線性離子阱互不干擾、平行工作的特點,使Orbitrap 專門進行一級掃描,線性離子阱專門進行二級DIA 掃描,線性離子阱掃描速度達20 Hz,比高分辨掃描更快。然后,使用一級超高分辨譜圖進行母離子色譜峰的精確提取和定量,二級譜圖只用于確證、不用于定量。因此,整體掃描速度明顯增加,同時二級DIA 掃描又無需關注掃描點數,為進一步縮小選擇窗口帶來可能。WiSIM-DIA 和Full MS-DIA 正是基于這種模式建立的DIA 技術(圖7)[56,57] 。WiSIM-DIA 將m / z 200 寬窗口一級選擇離子監(jiān)測掃描(Wide i-solation window-SIM)與m / z 12 窗口步長的二級離子阱全掃描相結合,24 萬分辨率的寬窗口SIM 掃描有效提高了母離子的選擇性,利用母離子精確質量提取色譜峰進行定量,相比經典DIA 的子離子定量靈敏度更高(圖7A)。同時,利用二級譜圖實現肽段定性確證。雖然與經典DIA 相比,WiSIM-DIA 二級為低分辨譜圖,但選擇窗口從m / z 25 縮小到了m / z 12,能有效減少基質背景的干擾。更進一步,Full MS-DIA 以m / z 3 步長進行二級采集,并插入若干24 萬分辨率的一級全掃描保證母離子掃描點數,與WiSIM-DIA 一樣,利用一級母離子定量、二級子離子定性(圖7B)。值得注意的是,Full MS-DIA 將選擇窗口縮小到僅m / z 3,其選擇性與DDA 相當,數據可以直接作為低分辨譜圖(母離子依1. 5 Da 質量精度)進行數據庫檢索,鑒定結果與DDA 高度吻合,首次使DIA 擺脫了依賴于譜圖庫的局限,初步實現了DIA與DDA 的統(tǒng)一[57] 。當然,DIA 數據直接進行數據庫檢索的算法和卡值方法也已出現,很快就會應用到DIA 中,從而使DIA 和DDA 一樣可以直接搜庫鑒定,徹底擺脫譜圖庫的限制,已有數據證明DIA 能夠獲得比DDA 更多的鑒定結果。
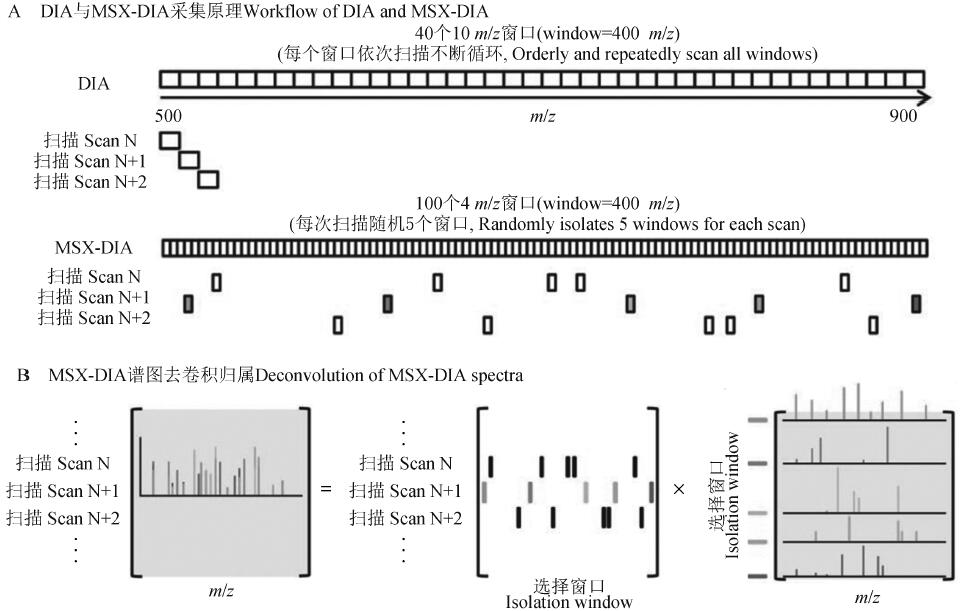
圖6DIA 與MSX-DIA 原理[54] :(A) DIA 與MSX-DIA 采集原理與比較; (B) MSX-DIA 譜圖通過去卷積歸屬獲得4 m / z 窗口譜圖
Fig. 6Schematic elucidation of data independent acquisition (DIA) and multiplexing-data independent acquisition (MSX-DIA)[54] : (A) Comparison of DIA and MSX-DIA; (B) Deconvolution of MSX-DIA spectra for 4 m / z selectivity
此外,最近發(fā)展的pSMART 技術,利用m / z 5 步長進行二級Orbitrap 掃描,并在掃描范圍內插入若干一級超高分辨Orbitrap 掃描,在Q-Orbitrap 平臺上實現了類似WiSIM-DIA 的采集技術[58] 。結果顯示,無論是定性還是定量分析,pSMART 的靈敏度、選擇性和重現性都要明顯優(yōu)于傳統(tǒng)DIA。
無論是相對定量還是絕對定量方法,DIA很好地克服了DDA鳥槍法和SRM目標監(jiān)測的種種不足, 在定量蛋白質組學中具有良好的應用前景。然而,目前DIA 方法的循環(huán)時間仍然較長,只能與納流液相聯(lián)用,并使用較長的梯度以獲得足夠的色譜峰寬,限制了DIA 的應用范圍。這也是DIA 技術下一步需要解決的問題。
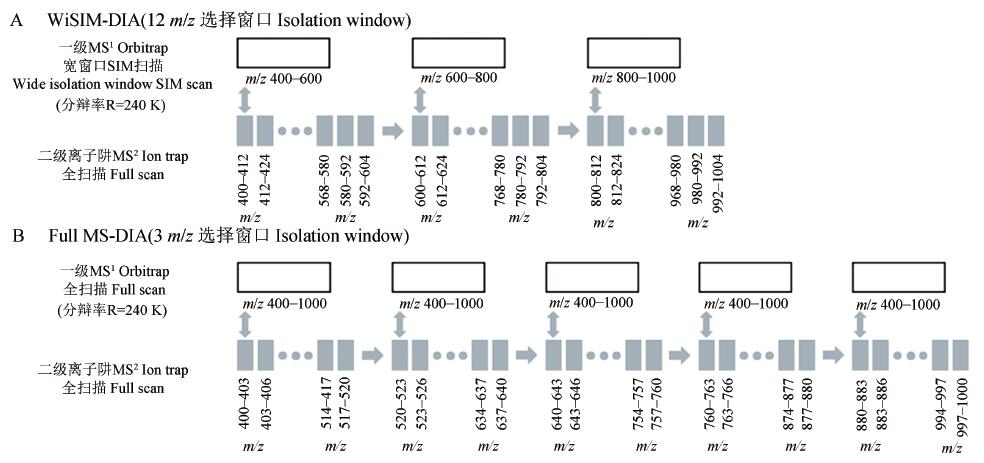
圖7WiSIM-DIA 與Full MS-DIA 原理[57] :(A) WiSIM-DIA 采集原理; (B) Full MS-DIA 采集原理
Fig. 7Schematic elucidation of wide isolation-SIM (WiSIM)-DIA and Full MS-DIA[57] : (A) Workflow of WiSIM-DIA; (B) Workflow of Full MS-DIA
5 總結與展望
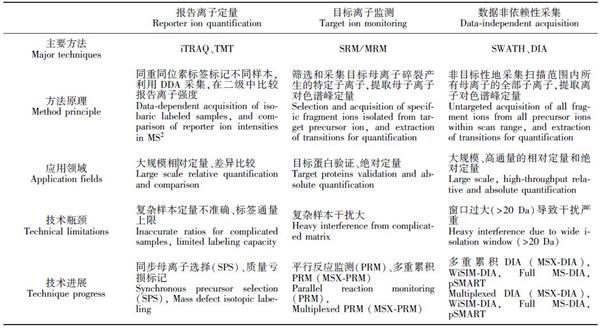
基于報告離子定量的同重同位素標記、目標離子監(jiān)測和數據非依賴采集已成為定量蛋白質組學的主要技術手段,表1 總結和比較了這3 種方法的原理和異同。定量蛋白質組學的飛速發(fā)展為質譜技術帶來挑戰(zhàn),基于穩(wěn)定同位素標記的相對定量和基于SRM 的絕對定量都面臨著復雜基質的嚴重干擾和通量不足等局限(表1)。而近來一系列高分辨質譜新技術的發(fā)展為解決這些問題帶來希望。其中,同步母離子選擇和質量虧損標記有效解決了相對定量的干擾和通量問題; 平行反應監(jiān)測及多重累積技術提高了SRM 的選擇性,成為絕對定量的新途徑; 數據非依賴性采集兼具DDA 與SRM 的優(yōu)勢,多重累積和三合一質譜技術使DIA 的掃描步長進一步縮小,能更有效地使DIA 技術應用于高通量的定量蛋白質組學。未來,這些新技術將逐漸取代傳統(tǒng)質譜技術,越來越多地應用到定量蛋白質組學中,為解決諸如蛋白質相互作用、臨床標志物研究等領域最棘手的問題帶來新的手段和突破。
表1 定量蛋白質組學主要技術的原理與進展
Table 1 Principle and progress of quantitative proteomic methods
References
1 Ong S E, Mann M. Nat. Chem. Biol. , 2005, 1(5): 252-262
2 Veenstra T D. J. Chromatogr. B, 2007, 847(1): 3-11
3 ZHOU Yuan, SHAN Yi -Chu, ZHANG Li -Hua, ZHANG Yu -Kui. Chinese Journal of Chromatography, 2013, 31(6):496-502
周愿, 單亦初, 張麗華, 張玉奎. 色譜, 2013, 31(6): 496-502
4 Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Anal. Bioanal. Chem. , 2007, 389(4): 1017-1031
5 ZHU Jin -Lei, ZHANG Kai, HE Xi -Wen, ZHANG Yu -Kui. Chinese J. Anal. Chem. , 2010, 38(3): 434-441
朱金蕾, 張鍇, 何錫文, 張玉奎. 分析化學, 2010, 38(3): 434-441
6 Lange V, Picotti P, Domon B, Aebersold R. Mol. Syst. Biol. , 2008, 4: 222
7 Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol. Cell. Proteomics, 2002, 1(5):376-386
8 Capelo J L, Carreira R J, Fernandes L, Lodeiro C, Santos H M, Simal -Gandara J. Talanta, 2010, 80(4): 1476-1486
9 Boersema P J, Raijmakers R, Lemeer S, Mohammed S, Heck A J. Nat. Protoc. , 2009, 4(4): 484-494
10 Koehler C J, Strozynski M, Kozielski F, Treumann A, Thiede B. J. Proteome Res. , 2009, 8(9): 4333-4341
11 Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed A K, Hamon C. Anal. Chem. , 2003, 75(8): 1895-1904
12 Ross PL, Huang Y N, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet -Jones M, He F, Jacobson A, Pappin DJ. Mol. Cell. Proteomics, 2004,3(12): 1154-1169
13 Karp N A, Huber W, Sadowski P G, Charles P D, Hester S V, Lilley K S. Mol. Cell. Proteomics, 2010, 9(9):1885-1897
14 Ow S Y, Salim M, Noirel J, Evans C, Rehman I, Wright P C. J. Proteome Res. , 2009, 8(11): 5347-5355
15 Ting L, Rad R, Gygi S P, Haas W. Nat. Methods, 2011, 8(11): 937-940
16 ZHAO Yan, YING Wan -Tao, QIAN Xiao -Hong. Chem. Life, 2008, 28(2): 210-213
趙焱, 應萬濤, 錢小紅. 生命的化學, 2008, 28(2): 210-213
17 Sherman J, McKay MJ, Ashman K, Molloy MP. Proteomics, 2009, 9(5): 1120-1123
18 Abbatiello SE, Mani DR, Keshishian H, Carr SA. Clin. Chem. , 2010, 56(2): 291-305
19 Kiyonami R, Schoen A, Prakash A, Peterman S, Zabrouskov V, Picotti P, Aebersold R, Huhmer A, Domon B. Mol. Cell. Proteomics, 2011, 10(2): M110. 002931
20 Cima I, Schiess R, Wild P, Kaelin M, Sch俟ffler P, Lange V, Picotti P, Ossola R, Templeton A, Schubert O, Fuchs T, Leippold T, Wyler S, Zehetner J, Jochum W, Buhmann J, Cerny T, Moch H, Gillessen S, Aebersold R, Krek W. Proc.Natl. Acad. Sci. USA, 2011, 108(8): 3342-3347
21 Picotti P, Bodenmiller B, Mueller L N, Domon B, Aebersold R. Cell, 2009, 138(4): 795-806
22 Pichler P, Kocher T, Holzmann J, Mazanek M, Taus T, Ammerer G, Mechtler K. Anal. Chem. , 2010, 82(15):6549-6558
23 Thingholm T E, Palmisano G, Kjeldsen F, Larsen M R. J. Proteome Res. , 2010, 9(8): 4045-4052
24 McAlister G C, Nusinow D P, Jedrychowski M P, W俟hr M, Huttlin E L, Erickson B K, Rad R, Haas W, Gygi S P. Anal.Chem. , 2014, 86(14): 7150-7158
25 Wuhr M, Haas W, McAlister G C, Peshkin L, Rad R, Kirschner M W, Gygi S P. Anal. Chem. , 2012, 84(21):9214-9221
26 Wenger C D, Lee M V, Hebert A S, McAlister G C, Phanstiel D H, Westphall M S, Coon J J. Nat. Methods, 2011,8(11): 933-935
27 Goeringer D E, Asano K G, McLuckey S A. Anal. Chem. , 1994, 66(3): 313-318
28 Viner R, Bomgarden R, Blank M, Rogers J. 61st ASMS, 2013, Poster W617
29 Blank M, Bomgarden R, Rogers J, Jacobs R, Fong J, Puri N, Zabrouskov V, Viner R. 61st ASMS, 2013, Poster Th449
30 Weekes M P, Tomasec P, Huttlin E L, Fielding C A, Nusinow D, Stanton R J, Wang E C, Aicheler R, Murrell I,Wilkinson G W, Lehner P J, Gygi S P. Cell, 2014, 157(6): 1460-1472
31 Dephoure N, Gygi S P. Sci. Signal, 2012, 5(217): rs2
32 Werner T, Becher I, Sweetman G, Doce C, Savitski M M, Bantscheff M. Anal. Chem. , 2012, 84(16): 7188-7194
33 McAlister G C, Huttlin E L, Haas W, Ting L, Jedrychowski M P, Rogers J C, Kuhn K, Pike I, Grothe R A, Blethrow J D, Gygi S P. Anal. Chem. , 2012, 84(17): 7469-7478
34 Werner T, Sweetman G, Savitski MF, Mathieson T, Bantscheff M, Savitski M M. Anal. Chem. , 2014, 86(7): 3594-3601
35 Gallien S, Duriez E, Demeure K, Domon B. J. Proteomics, 2013, 9(81): 148-158
36 Karlsson C, Malmstrom L, Aebersold R, Malmstrom J. Nat. Commun. , 2012, 3: 1301
37 Gallart -Ayala H, Moyano E, Galceran M T. J. Chromatogr. A, 2008, 1208(1 -2): 182-188
38 Mart侏nez -Villalba A, Moyano E, Martins C P, Galceran M T. Anal. Bioanal. Chem. , 2010, 397(7): 2893-2901
39 Fortin T, Salvador A, Charrier J P, Lenz C, Bettsworth F, Lacoux X, Choquet -Kastylevsky G, Lemoine J. Anal. Chem. ,2009, 81(22): 9343-9352
40 Peterson A C, Russell J D, Bailey D J, Westphall M S, Coon J J. Mol. Cell. Proteomics, 2012, 11(11): 1475-1488
41 Schiffmann C, Hansen R, Baumann S, Kublik A, Nielsen P H, Adrian L, von Bergen M, Jehmlich N, Seifert J. Anal.Bioanal. Chem. , 2014, 406(1): 283-291
42 Gallien S, Duriez E, Demeure K, Domon B. J. Proteomics, 2013, 81: 148-158
43 Tsuchiya H, Tanaka K, Saeki Y. Biochem. Biophys. Res. Commun. , 2013, 436(2): 223-229
44 Tang H, Fang H, Yin E, Brasier A R, Sowers L C, Zhang K. Anal. Chem. , 2014, 86(11): 5526-5534
45 Gallien S, Bourmaud A, Kim S Y, Domon B. J. Proteomics, 2014, 100: 147-159
46 Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Mol. Cell. Proteomics, 2012, 11(12): 1709-1723
47 Law K P, Lim Y P. Expert. Rev. Proteomics, 2013, 10(6): 551-566
48 Venable J D, Dong M Q, Wohlschlegel J, Dillin A, Yates J R. Nat. Methods, 2004, 1(1): 39-45
49 Gillet L C, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Mol. Cell. Proteomics, 2012,11(6): O111. 016717
50 Liu Y, Huttenhain R, Surinova S, Gillet L C, Mouritsen J, Brunner R, Navarro P, Aebersold R. Proteomics, 2013,13(8): 1247-1256
51 Collins B C, Gillet L C, Rosenberger G, Rost H L, Vichalkovski A, Gstaiger M, Aebersold R. Nat. Methods, 2013, 10(12): 1246-1253
52 Lambert J P, Ivosev G, Couzens A L, Larsen B, Taipale M, Lin Z Y, Zhong Q, Lindquist S, Vidal M, Aebersold R,Pawson T, Bonner R, Tate S, Gingras A C. Nat. Methods, 2013, 10(12): 1239-1245
53 Chapman J D, Goodlett D R, Masselon C D. Mass Spectrom. Rev. , 2013: 10. 1002/ mas. 21400
54 Egertson J D, Kuehn A, Merrihew G E, Bateman N W, MacLean B X, Ting Y S, Canterbury J D, Marsh D M, Kellmann M, Zabrouskov V, Wu C C, MacCoss M J. Nat. Methods, 2013, 10(8): 744-746
55 Senko M W, Remes P M, Canterbury J D, Mathur R, Song Q, Eliuk S M, Mullen C, Earley L, Hardman M, Blethrow JD, Bui H, Specht A, Lange O, Denisov E, Makarov A, Horning S, Zabrouskov V. Anal. Chem. , 2013, 85(24):11710-11714
56 Kiyonami R, Patel B, Senko M, Zabrouskov V, Egertson J, Ting S, MacCoss M, Rogers J, Huhmer A. Large Scale Targe -ted Protein Quantification Using WiSIM -DIA Workflow on a Orbitrap Fusion Tribrid Mass Spectrometer. ASMS, 2014, W737
57 ZHANG Wei, Reiko Kiyonami, JIANG Zheng, CHEN Wei. Chinese J. Anal. Chem. , 2014, 42(12): 1750-1758
張偉, Reiko Kiyonami, 江崢, 陳偉. 分析化學, 2014, 42(12): 1750-1758
58 Prakash A, Peterman S, Ahmad S, Sarracino D, Frewen B, Vogelsang M, Byram G, Krastins B, Vadali G, Lopez M. J. Proteome Res. , 2014, doi: 10. 1021/ pr5003017
Progress in Mass Spectrometry Acquisition Approach for Quantitative Proteomics
ZHANG Wei*
(Thermo Fisher Scientific, Shanghai 201206, China)
Abstract Mass spectrometry is an important and powerful tool for protein quantification. With the in -depth development of quantitative proteomics, limitations of classic MS based quantification methods, such as complicated matrix interference and throughput/ capacity limitation, start to appear. Recent progress of series novel MS based techniques provide effective solutions for the limitations of relative and absolute proteomic quantification, including synchronous precursor selection ( SPS), mass defect isobaric labeling, parallel reaction monitoring (PRM), multiplexing acquisition (MSX), and various novel data independent acquisition(DIA) modes. Here we summarized the current limitations of quantitative proteomics, reviewed the latest MS based quantification approaches, and discussed the features and advantages of these novel techniques for quantitative proteomic application.
Keywords Quantitative proteomics; Synchronous precursor selection; Parallel reaction monitoring; Data independent acquisition; Review
(Received 10 September 2014; accepted 18 October 2014)



Copyright(C) 1998-2024 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com