
��(d��ng)ǰλ�� > ��� > ���g(sh��)���� > ���ڔ�(sh��)��PCR�Ćη���DNA�������g(sh��)�о��M(j��n)չ
���ڔ�(sh��)��PCR�Ćη���DNA�������g(sh��)�о��M(j��n)չ
���ڔ�(sh��)��PCR�Ćη���DNA�������g(sh��)�о��M(j��n)չ
����1)��־��1)����1)갳�1)��Ц��2)
��1)�Ї�Ӌ(j��)���ƌW(xu��)�о�Ժ������100013 ��2)�㽭ʡӋ(j��)���ƌW(xu��)�о�Ժ������310013)
ժҪ ��(sh��)��PCR��һ�(xi��ng)ᘌ��η���Ŀ��(bi��o)DNA�Ľ^���������g(sh��)��ԓ���g(sh��)�nj�����DNAģ��ķ���(y��ng)��Һ���䵽������(d��)���ķ���(y��ng)���в��Ұl(f��)���U(ku��)������(y��ng)��ͨ�^�y(t��ng)Ӌ(j��)����(y��ng)���е������̖������DNA�Ŀ�ؐ��(sh��)��DNA��Ʒ�ڷ���(y��ng)�����S�C(j��)�ͪ�(d��)���ֲ��džη��ӳɹ��U(ku��)���͜�(zh��n)�_����D NA��ؐ��(sh��)���P(gu��n)�I���������ľC���˔�(sh��)��PCR�İl(f��)չ�vʷ����(sh��)��PCR�c��(sh��)�r(sh��)�ɹⶨ��P CR�ą^(q��)�e���Լ���(sh��)��PCR���R���\�����D(zhu��n)����ɷֶ������μ�(x��)��������_(d��)���h(hu��n)������z�y����һ���y��ȷ���������M(j��n)չ����չ����ԓ���g(sh��)�đ�(y��ng)��ǰ��
�P(gu��n)�I�~ ��(sh��)��PCR���^����������(sh��)�r(sh��)�ɹⶨ��PCR����ؐ��(sh��)���η��ӡ�
����������������Ժ��S�ȷ����ǬF(xi��n)������W(xu��)�Ļ��A(ch��)�����ᶨ�����g(sh��)���\��͂�(g��)�w���t(y��)����ʳƷ�z�(y��n)���D(zhu��n)��������z�y����ԭ�w�b�������t(y��)�ƌW(xu��)�ȷ����ѱ��V����(y��ng)����ͬ�r(sh��)�@Щ��(y��ng)��Ҳ�Ƅ��˺��ᶨ�����g(sh��)���M(j��n)����
DNA���ӔU(ku��)�����g(sh��)�đ�(y��ng)�ô��M(j��n)�˷�������W(xu��)���g(sh��)�İl(f��)չ[2]�����_���� DNA ��ؐ��(sh��)�ǬF(xi��n)����������W(xu��)���t(y��)�W(xu��)����Ҫ��(y��ng)��֮һ����(sh��)��PCR(digital polymerase chain reaction��dPCR)���� DNA �������¼��g(sh��)���˷��ˌ�(sh��)�r(sh��)�ɹⶨ��PCR (real timequantitative PCR��qPCR)��һϵ��ȱ�c(di��n)����(sh��)�F(xi��n)�ˆη��� DNA �^��������dPCR�nj���(g��) DNA ��Ʒ����(y��ng)Һ�քe�M(j��n)�Д�(sh��)��Ӌ(j��)�ķ���(y��ng)������ÿ��(g��)����(y��ng)�քe�M(j��n)�ДU(ku��)���z�y��dPCR�� DNA �^��������������Q��qPCR ���õĘ�(bi��o)��(zh��n)�������y���Y(ji��)���a(ch��n)��Ӱ푵Ȇ��}�����ɜp�� qPCR �����Ļ��wЧ��(y��ng)���˼��g(sh��)���R���\�ࡢ�D(zhu��n)����ɷֶ������μ�(x��)��������_(d��)���h(hu��n)������z�y����һ���y��ȷ�����о��l(f��)�]����Ҫ������
1 dPCR���g(sh��)�l(f��)չ�vʷ
1992����Sykes �Ȉ�(b��o)���˻��ژ�ƷϡጺͲ��ɷֲ���(sh��)��(j��)̎���ij�ʽPCR�������g(sh��)��������˔�(sh��)��PCR�Ę�(g��u)�롣1997����Kalinina ��ʹ�ò���ë��(x��)���M(j��n)������(��l) ����PCR����(y��ng)��ͨ�^�ɹ�̽��ռ�������MDNA�Ćη�����̖���M(j��n)���l(f��)չ�ˆη��Ӷ������g(sh��)��1999��, Vogelstein ��Kinzler�������ψ�(b��o)������96�װ�ϵ�y(t��ng)�l(f��)չ��������PCR�U(ku��)�����������ڽY(ji��)�c�����°�ͻ׃����ras �Ķ����������S֮��Dressman �Ȱl(f��)����һ�NBEAMing����( ���ӡ��鄩���U(ku��)���c����)�����ù�H�����c���鹲�r(ji��)�Y(ji��)�������������һ�������ص�PCR����M(j��n)�ДU(ku��)������(y��ng)�����黯���ú���ʹ����ʽ��(x��)���xͨ�^���������λ����׃�����M�ܲ��E�^����ԓ���g(sh��)�Բ�ʧ��V����(y��ng)���ڌ�(sh��)�(y��n)�Ҷ���DNA�����뷽���������Á�z�y�Ͷ����κ�������B(t��i)��(single nucleotide polymorphism��SNP) ��ͻ׃�Ⱥ�������׃��������׃����λ����Ĵ����܉����w���x��������ɑ�(y��ng)���ژ�Ʒ�y������з�����
�M��dPCR���g(sh��)�߂�V韑�(y��ng)��ǰ����������96��384 ��ƽ��Әӵď�(f��)�s�����龫�_�y�����������y��Ҳ�y�Խ�Q��ͨ���y�����}�����w�ij��F(xi��n)�ͼ{��(nl) ����(y��ng)�x�����_�l(f��)�˷����@Щ���g(sh��)��ƿ�i����2003�꣬Liu��ʹ�����w�ĸ����������wоƬ���M(j��n)����400 ��(g��)��(d��)����3nl PCR����(y��ng)��2008 ����Ƕ��ʽоƬ��PCR ����(y��ng)�Δ�(sh��)�_(d��)����9180 ��(g��)6nl ��PCRƽ�з���(y��ng)��������F(xi��n)luidigm��˾��Biomark ϵ�y(t��ng)����ϵ�y(t��ng)���wdPCRоƬ��12��(g��)��(d��)�������w���(panel) ������(y��ng)�M(j��n)�ӿM��(�D1). ÿ��(g��)��庬��765 ��(g��)6nl �Ī�(d��)������(y��ng)��(partition)��4.6��l����(y��ng)Һͨ�^�M(j��n)�ӿ��M(j��n)����䵽765��(g��)����(y��ng)���M(j��n)�Ъ�(d��)���U(ku��)����ԓ���g(sh��)����څ���Ƶ�ͬ�r(sh��)���δεķ���(y��ng)��(sh��)�͙z�y���ȳɞ���(xi��ng)���g(sh��)�о������c(di��n)�������Heyries �Ȉ�(b��o)����һ��(g��)���f�������wdPCR�b�����ɞ���dPCR���g(sh��)����һ�ش�ͻ�ơ�ԓ�b��ͨ�^���揈�����_(d��)����Ʒ�ֲ�����ˮ���ƣ�����ˆη���DNA�U(ku��)���ı�������ÿƤ��( pl) �M(j��n)��100�f��(g��)����(y��ng)���_(d��)��ÿƽ������(cm2) 44�f��(g��)����(y��ng)���@�N�b�ÿɌ�(sh��)�F(xi��n)ÿ10�f��(g��)Ұ���������е���һ��(g��)׃����ؐ�ęz�y��

�D1 ���wdPCR��12��765 ����(y��ng)оƬ
2 qPCR�cdPCR���^�о�
qPCR��ĿǰDNA�����о�����Ҫ���g(sh��)��ԓ���g(sh��)ͨ�^��PCR����(y��ng)�wϵ�м���ɹ�Y(ji��)��Ⱦ��(SYBR green I) ��ɹ��(bi��o)ӛ��̽�( ��TaqMan Probes) �����Ì�(sh��)�r(sh��)�e�۵ğɹ���̖�O(ji��n)�y����(g��)�U(ku��)���^�������ͨ�^��(bi��o)��(zh��n)������δ֪ģ���M(j��n)�ж����������Դˁ��u��PCR�ĔU(ku��)��Ч����qPCR��Ҫ��ه��У��(zh��n)���Ƃ�Ę�(bi��o)��(zh��n)�������M(j��n)���_��δ֪��Ʒ�ĝ�ȣ������һ�N���������ķ�����ԓ���g(sh��)���������}��a��У��(zh��n)Ʒ�͘�Ʒ�g�����IJ�ͬ������ƫ��������Ӱ�PCR��Ч�ʺ͜y��푑�(y��ng)��b���Ϳ�ؐ��(sh��)��Ŀ��(bi��o) DNA���Ӳ���ͨ�^�U(ku��)���z�y����c����Ʒ��PCR�U(ku��)��Ч�ʿ����cУ��(zh��n)��ĔU(ku��)��Ч�ʲ�ͬ��d��DNA��ȡ�r(sh��)����DNA��Һ���s�|(zh��)����DNA����Ӱ���PCR�ӑB(t��i)�U(ku��)���^����
dPCR��һ�(xi��ng)�z�y�Ͷ���������¼��g(sh��)������ͬ�ڂ��y(t��ng)��qPCR����?y��n)����ֱ���?j��)��(sh��)Ŀ��(bi��o)���Ӕ�(sh��)���������κ�У��(zh��n)������(bi��o)��dPCRͨ�^Ӌ(j��)��(sh��)��(g��)���ӏĶ���(sh��)�F(xi��n)�^��������dPCR�����y(t��ng)PCR��ָ��(sh��)��(sh��)��(j��)�D(zhu��n)�Q�ɔ�(sh��)����̖���H�Hͨ�^�@ʾ�����O(sh��)����ѭ�h(hu��n)��(sh��)��U(ku��)���Ƿ�l(f��)�������ɿ˷��������y���_(d��)������Ľ^��������dPCR���M(j��n)�ДU(ku��)������(y��ng)ǰ��������DNAģ���PCR��Һϡጺ�ֲ��������Ī�(d��)������(y��ng)��(�D2) ���η����gͨ�^ϡጷ��x�����Ҫ�(d��)���M(j��n)��PCR�U(ku��)����������ÿ��(g��)�U(ku��)���a(ch��n)�����@��(sh��)�F(xi��n)��ͨ�^����PCR�U(ku��)���Ę�Ʒ���x���@�N��Ʒ�ķ����������������̖��Ӱ�����ߵ� �S�ȰИ�(bi��o)�ĔU(ku��)���`����������Ӌ(j��)���DNA��ģ�忽ؐ��(sh��)������Ҫ���Å�����(bi��o)��(zh��n)��������(bi��o)����(zh��n)�_�Ķ�����ه��40��45��(g��)ѭ�h(hu��n)�ĔU(ku��)�����e(cu��)�`����ԙz��ˮƽ(��DNAģ����F(xi��n)�ڷ���(y��ng)�Ҷ�δ���z��) �dz��͡���(sh��)��PCR��һ�(xi��ng)���п��؏�(f��)�ԵĶ�����DNA���ӵă�(y��u)�����g(sh��)��������������z�yͨ���ߡ��خ��ԏ�(qi��ng)���`���ȸ���������(zh��n)�_�ȃ�(y��u)�c(di��n)ʹ��ɞ��˷�������W(xu��)�о��е���Ҫ������
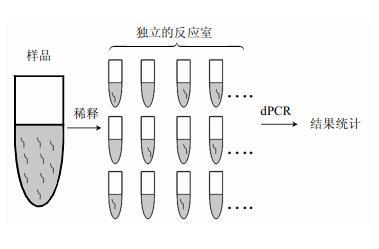
�D2 dPCR�U(ku��)������(y��ng)ԭ��
3 dPCR��(y��ng)���о�
3.1 dPCR���R���\�����о�
dPCR��(y��ng)���ڰ��Y�ĵ�λ����ͻ׃�Ϳ�ؐ��(sh��)׃���ȷ���ęz�y�о����鰩�Y�z�y�ṩ���µ��\��ߡ�Oehler �Ȍ�����Ѫ�������P(gu��n)����ABL�Ұ��ἤø�Y(ji��)��(g��u)��ͻ׃������dPCR�M(j��n)�н^�������z�y�����ҽ�����ͬ�r(sh��)�z�y����(g��)ABL�Ұ��ἤø�Y(ji��)��(g��u)��ͻ׃�ķ�������Ƥ���L�������w( epithelial growth factor receptor ��EGFR) ͻ׃���^���_(d��)һ������l(f��)�[����Yung ���_�l(f��)��һ�N�P(gu��n)�ڷ�С��(x��)���Էΰ�����Ѫ�{���[���еăɷNEGFRͻ׃�w( ��19���@�Ӄ�(n��i)ȱʧ�͵�21���@��L858R ͻ׃) ��dPCR�����z�y��������19���@�Ӄ�(n��i)ȱʧ�͵�21���@��L858Rͻ׃ռ�Ұ��ἤøͻ׃푑�(y��ng)��85% ���ϡ���(sh��)�(y��n)�Y(ji��)�������@�ɷNͻ׃���A(y��)�ί���35��(g��)Ѫ�{��Ʒ�քe�z��6��(g��)(17%)��9��(g��)(26%)���c�[����(x��)���Ĝy��Y(ji��)�������Ѫ�{EGFRͻ׃���`���Ⱥ��خ�����92% ��100%�����[����Ѫ�{�еĵ��S��EGFRͻ׃ͨ�^���wdPCR�`���z�y�;��_��������(y��ng)�����A(y��)�y�ί�����(y��ng)���O(ji��n)�ؼ����M(j��n)չ�����ڙz�y�@������ˎ�ȷ����о���
��ͨ����������(y��n)�C�c�������P(gu��n)��SNPs�о���ʹ�о��߲���һ�(xi��ng)�µļ��g(sh��)��dPCR��������ѪҺ����ǻϴҺ��ȡDNA�Ļ�������M(j��n)���u�r(ji��)���������û���BioMark PCRƽ�_��Fluidigm 48��48�ӑB(t��i)�������оƬ�M(j��n)��SNP�����������90��(g��)��Ʒ�c20��(g��)SNP���wϵ�������քe�õ�ƽ��99.7%�Լ�16��(g��)�wϵ100% �ĽY(ji��)����TaqMan ��������c���MSNP�M(j��n)�Ќ��ȵõ���100%�����P(gu��n)���������о�DNAģ��׃����Ӱ���ѪҺ��Ʒ( CH-1��n=20��CH-2��n=47��KK��n=47)�Ϳ�ǻϴҺ( n=37) ͨ�^24��(g��)SNPs �M(j��n)�л��������CH-1 ��CH-2 �����@ʾ�˺ܺõĻ���푑�(y��ng)(��98.8%)��KK���κͿ�ǻϴҺ��Ʒ푑�(y��ng)�^��(82.1% ��94.0 %)������(j��ng)�ټ������KK�Ϳ�ǻϴҺ��Ʒ�Y(ji��)��푑�(y��ng)�����@�����( ��98.8% ).�о��Y(ji��)������dPCR�Ĝ�(zh��n)�_���cTaqMan ��������������DNA������������(ji��)ʡ�˸�����������r(sh��)�g���ɱ���
��ؐ��(sh��)׃��(copy number variation��CNV) ������z��׃������Ҫ��Դ�������c������������������P(gu��n)��Qin�Ȳ���dPCR ϵ�y(t��ng)���_������DNA��Ʒ�Ŀ�ؐ��(sh��)��������dPCR�ķ���ģ�ͣ������M(j��n)��������3��(g��)������о���a����һϵ�������M�ͺϳɵ�RPP30�����ж�����RPP30����Ŀ�ؐ��(sh��)���c�A(y��)�ڽY(ji��)�������b���о�����Ʒ����Ʒ�е�CYP2D6����ؐ��(sh��)���c���y(t��ng)�ֶζ��x�Ŀ�ؐ��(sh��)һ����c��ͨ�^��ERBB2 ����ĔU(ku��)���Y�x����40��(g��)���ٰ���Ʒ���c�īI(xi��n)��(b��o)���Ļ���һ�����@�(xi��ng)�о�������dPCR���خ���CNV ���о�����һ��(g��)�µ������_�ĺ͏�(qi��ng)�����ļ��g(sh��)֧����
�ЋDѪ�{�е�̥�����x����(DNA��RNA�z�y�_���˟o��(chu��ng)�a(ch��n)ǰ�\������I(l��ng)�����a(b��)�������ϾC�ϰY(��������Ⱦɫ�w�����w) �a(ch��n)ǰ�\����������̥������ľ��_dPCR�������M(j��n)�˴��о��I(l��ng)��İl(f��)չ�����ЋD��̥���ṩ����ȫ�\������2007����Lo�����C����dPCR �������ЋDѪ�{RNA- SNP��λ������ʵĜy��������(y��ng)��ԓ�����������w̥�PDNA ��Ʒ���b�e��21��(g��)�����w��Ʒ��Lo���M(j��n)һ���C������ʹ�����wDNA��С��Ƭ�δ��ڕr(sh��)�������wҲ�ܱ��z�y�������ߌ���ᘌ�ĸ�HѪ�{�����������w�z�y��ʽ��ʮ�ֱ�Ҫ����Fan��Quake���m(x��)�l(f��)�����īI(xi��n)Ҳ֧�����@��(g��)�^�c(di��n)���S��Lo��ᘌ��Ե����|(zh��)- RNA ��(f��)���﷽ʽ������Ѫ�{�е�̥�����xRNA�M(j��n)�����о������^���|(zh��)�V��dPCR���ЋDѪ�{��PLAC4�����mRNA-SNP���ʣ������|(zh��)�V���R���`���Ȟ�90%���R���خ��Ԟ�96.5%��dPCR���R���`���Ⱥ��خ��Ծ���100%�������о��������c�|(zh��)�V�z�y��qPCR�����dPCR���ԫ@�ø����_�Ķ����Y(ji��)�����ʴˣ�dPCR�z�y̥��DNA�ĝ�ȿ���Ҫ�����A(y��)�������wоƬdPCR�Ŀ��ٰl(f��)չ���ѳɞ�Ŀǰ�R����(y��ng)����ߝ����ğo��(chu��ng)�a(ch��n)ǰ�\�༼�g(sh��)���������ڳ�Ҏ(gu��)�z���еđ�(y��ng)�õ춨�˻��A(ch��)��
3.2 dPCR���D(zhu��n)����ֲ��z�y������о�
�D(zhu��n)����ֲ�P���P(gu��n)ʳƷ�Ķ���������Ҫ�y���D(zhu��n)����������������Ŀǰ����qPCR������ᶨ��������dPCR���Բ���ҪУ��(zh��n)�����(zh��n)�_�y���Ϳ�ؐ��DNA������Corbisier ����dPCR��������ȡ��MON810���N�ӵ���Դ�z�y�����hmg����Ŀ�ؐ��(sh��)���(y��n)�C��dPCR�Ľ^����ؐ��(sh��)���������Y(ji��)�����|(zh��)��DNA����(bi��o)��(zh��n)���|(zh��)qPCR������������һ����������dPCR����Ӌ(j��)�������������Á�y���D(zhu��n)�������P(gu��n)��(bi��o)��(zh��n)���|(zh��)��D NA��ؐ��(sh��)���ʡ�
�η��ӔU(ku��)��Ч�ʌ��ڸ��ƿ�DNAƬ�εĿ�ؐ��(sh��)�Ͷ�Ƭ������DNA�Ĺ�Ӌ(j��)ƫ�����@�����x��Ŀ��(bi��o)DNA�����ڷ���(y��ng)�ҵ��S�C(j��)�ͪ�(d��)���ֲ���dPCR��(zh��n)�_�������P(gu��n)�I��Bhat ���J(r��n)��������(y��ng)�ҵ������Dz��_���ȵ���Ҫ��Դ�������u�����������_������6%���¡��@�(xi��ng)�l(f��)�F(xi��n)������������dPCR�y��������ˮƽ�о����S��Burns �Ȳ��ý�(j��ng)�(y��n)�C�ęz�y�D(zhu��n)����ɷֵ�qPCR����(y��ng)�wϵ���u����dPCR���g(sh��)�Ľ^���z�y�Ͷ�������Ҳ�U���˲����ݶ��A(y��)��(sh��)�(y��n)�ķ�ʽ����ʹdPCR�����_�y����ؐ��(sh��)����(j��ng)�^30��(g��)ƽ���؏�(f��)��(sh��)�(y��n)�Y(ji��)��������ÿ��(g��)ƽ��l(f��)��200 ��700��(g��)����(y��ng)����龫�_�����@�̓x���S�̵����]��һ�µ���
3.3 dP CR�چμ�(x��)��������_(d��)������о�
��dPCR ���g(sh��)���چμ�(x��)��������_(d��)�о���dPCR���g(sh��)�l(f��)չ����̱���White ���O(sh��)Ӌ(j��)�_�l(f��)��һ�N�܉�߾��Ȝy���μ�(x��)���Д�(sh��)��Ӌ(j��)�Ļ�����_(d��)��dPCR�O(sh��)�������O(sh��)����M(j��n)�а�����(x��)���@ȡ���ѽ������D(zhu��n)䛺Ͷ���PCR�Ȇμ�(x��)���ӹ�̎�����������������ͨ�������ͳɱ���White �Ȳ������w�e̎��p���˜y���������������`����������ˆκ�������خ��ԡ����ґ�(y��ng)���@�(xi��ng)���g(sh��)�y����3300��(g��)�μ�(x��)����������a��miRNA��K562 ��(x��)���ı��_(d��)��b��miRNA����Ŀ��(bi��o)�D(zhu��n)�������̥��(x��)���ֻ��r(sh��)�ąf(xi��)ͬ���ã�c�����ٰ���(x��)���Ćκ���ͻ׃�z�y���������ĺ��Ĺ����ṩ�˶�N����оƬϵ�y(t��ng)�Ćμ�(x��)���D(zhu��n)䛷�����
Spurgeon�Ȉ�(b��o)���˻������wdPCR�ĸ�ͨ��������_(d��)ƽ�_����ƽ�_ͬ�r(sh��)��2304��(g��)�����M(j��n)�ДU(ku��)������(y��ng)���c96�װ������Ҫ���ٵ��Ϙ������چ�оƬ��߀���ԙz�y18��(g��)�M���е�45��(g��)��ͬ��������(sh��)�(y��n)�Y(ji��)���c���y(t��ng)��qPCRʮ���Ǻ�������ͬ�̘I(y��)����DNAоƬ��Ⱦ��и��õ��؏�(f��)����Warren�Ȉ�(b��o)���˔�(sh��)��RT-PCR���wоƬ�Ɍ�(sh��)�F(xi��n)�D(zhu��n)����ӱ��_(d��)��(sh��)����ϵ�y(t��ng)�������������ҿ���Ӌ(j��)��Դ�Ԇμ�(x��)����cDNA��Ʒ����һ��(g��)��������5 ����Ѫǰ�w���о�����Warren �l(f��)�F(xi��n)��Ѫ�ɼ�(x��)�����D(zhu��n)�����PU1���_(d��)�@��׃������flk2-��flk2+�����漚(x��)����PU1���_(d��)�ʶ����ԡ�
�����Guo�Ȳ���dPCR���g(sh��)�о����ܾ��ѵ����ߵĆμ�(x��)��������_(d��)�����Æμ�(x��)�����_(d��)��������С��������64��(x��)����3 �N��ͬ��(x��)����͵�500��(g��)��(x��)���ֻ������A���M(j��n)�����о�������800����(g��)�D(zhu��n)��ӵ�ȫ��̥�������x����48��(g��)�����M(j��n)���о����Y(ji��)���l(f��)�F(xi��n)3 �N��(x��)����Ϳ������ö������_(d��)�����M(j��n)�����@�ą^(q��)�֡����քe�_��ID2��Sox2�鼚(x��)����ͼ�(x��)����(n��i)����Ę�(bi��o)ӛ�����M(j��n)�������ڃ�(n��i)��(x��)�����w��ό�Fgfr2/Fgf4 �M(j��n)��ؓ(f��)���P(gu��n)���_(d��)��������λ�c��̖���f�l(f��)�����D(zhu��n)䛻�ӳ���֮ǰ���Y(ji��)�������μ�(x��)�����_(d��)�����܉������о���(x��)���İl(f��)���C(j��)�ƣ���(y��ng)���@�(xi��ng)���g(sh��)�ƏV��(y��ng)���ڸ��V��������ϵ�y(t��ng)����
3.4 dP CR�ڭh(hu��n)�����﷽����о�
dPCR���g(sh��)��ͨ���U(ku��)���ͷ����ă�(y��u)�c(di��n)ʹͬ�r(sh��)�о���Ȼ��Ķ���(g��)��(x��)���μ�(x��)���Ķ���(g��)��ͬ������Ԍ�(sh��)�F(xi��n)��Ottesen ��ʹ��һ��(g��)�ڰ�ρ�����c����Ⱥ���P(gu��n)�Ķ༉�P(gu��n)�Iø���������T�D������dPCR���g(sh��)�l(f��)�F(xi��n)��δ֪�ĺ����wRNA���@�N���g(sh��)�܉�ϵ�y(t��ng)���b����(f��)�s���B(t��i)�h(hu��n)���Дy��Ŀ�Ļ���ļ�(x��)����������(j��)һ�ɂ�(g��)Ŀ�Ļ����ݵ���N���������dPCR����h(hu��n)���о��I(l��ng)���ṩ�µ����C(j��)��
���������ǵ����ϴ���������������N��Ȼ�������ڴ����(sh��)�����ļ������s�r�л���M�W(xu��)��(j��ng)����A�΄��ּ��g(sh��)�����M(j��n)���о���Tadmor �Ȳ���dPCR������ͨ�^һ��(g��)������(bi��o)־����?q��)��μ?x��)����(x��)���c��Ȼ�h(hu��n)�����P(gu��n)(li��n)����(y��ng)���@�(xi��ng)���g(sh��)����ρ���c����Ⱥ����о��l(f��)�F(xi��n)�ڌٵķ�����Ⱦģʽ�g����������Čك�(n��i)�x���ԡ�������(bi��o)־���@ʾ�������g��(y��n)��ĵ�λ����������ʾ�˳�����������ĵ�λ����M������D(zhu��n)�Ƶ����Ƴ̶��������o�����B(y��ng)������(x��)���������S��h(hu��n)����(x��)���c������������ṩ����Ч������
3.5 ����dPCR�Ćη��Ӝy���g(sh��)�о�
�ԏ�2003��η��Ӝy���g(sh��)��(y��ng)�õ��y���I(l��ng)���������g(sh��)�ĸ����ʽ���^��(b��o)����������ÿ��10��ͨ���������L��
dPCR���ͨ���y���ṩ���`���ĺͽ^����У��(zh��n)����һ���y��(next generation sequencing, NGS)��454��Solexa ��SOLiDƽ�_��Ҫͨ�^�y��У�����Ӕ�(sh��)�����˗l�����ڃɷN���������a�������˼��Ę�Ʒ��Ҫ�Ƃ���Ď죬��������˿ɜy���Ʒ�ķ�����������(sh��)��(y��ng)�Á��v�����������M�����ŌW(xu��)��Ʒ�����t(y��)��Ʒ���R����Ʒ�y����DNA�Ę�Ʒ���Ƿdz�������b��ÿ��(g��)�Ď���Ҫ�ζ����y������������˳ɱ�, �����˜y���ͨ��. ���, ʹ��dPCR���_����454 ��Solexa �y���Ď���ʹ�y���Ď���Ƃ��_(d��)���{�˼���ͬ�r(sh��)�����˻��M(f��i)�ڃx���ζ��ijɱ��͕r(sh��)�g��ͬ�r(sh��)�ɹ��،�454 FLX ��Sole xa�y��ƽ�_�ĵ��ڼ{�˼��ļ�(x��)���Ͳ�������DNA��Ʒ�M(j��n)���˜y����White �ȵ��о��Y(ji��)���״����_�C���˻���454 ƽ�_��Ƥ�˼�DNA��Ʒ�Ĝy������DNA��Ʒ���������ڟo�A(y��)�U(ku��)���r(sh��)������1000 ���dPCR��(sh��)�F(xi��n)�˜y���Ď�Ľ^�������������˘�(g��u)����PCR������(bi��o)��(zh��n)�����Ȳ��_��������������(bi��o)��(zh��n)ƫ�����10% ���ڟo�ζ���r�������M��ֱ�Ӝy��ľ���Ҫ��
ABCA4����ͻ׃���λ����һ�¿������N������Zernant �Ȟ錤��ABCA4���������P(gu��n)׃���������о���168 ���t(y��)�W(xu��)�\�����S�߲�׃��ҕ�F-ҕ�U�I�B(y��ng)����������ABCA4�������͵Ļ���������ʹ��ABCA4оƬ�Y�x����ͻ׃���Y(ji��)���l(f��)�F(xi��n)111 ����������1 ��������2 ��(g��)�A(y��)�ڵ�ͻ׃��57��(g��)�����Л]�У��S��(y��ng)��dPCR��454 �y��ƽ�_�b������׃���Y(ji��)�����������@Щ�²��Ե�ԭ���������b����һλ�c(di��n)ͻ׃��103 �����������b���˵ڶ��������P(gu��n)��λ����49�����mȻ���a ABCA4����ı���ͻ׃?n��i)�Ȼδ֪�������S��λ��ABCA4�ķǾ��a�^(q��)������ABCA4��(sh��)�(y��n)�Y(ji��)����һ��(g��)�^�õ��x����NGS ƽ�_���ںY�x�����ļ�����һ��(g��)�ڕr(sh��)�g�����M(f��i)���涼��Ч�Ĺ�����
NGS ��һ�N�R�e�ʹ_�J(r��n)δ֪�²�����ǰ���V韵ļ��g(sh��)��Ȼ�����������������������(y��ng)�õȷ���ĕr(sh��)Ч�ԣ��s������?y��n)�ȱ����������Ч���ɿ����Ԅ�DNA��Ʒ�Ƃ䷽�����ܵ����ơ�����ͻ���@�N������Kim ���O(sh��)Ӌ(j��)��һ�N�������w�ֲ�Ԫ���Ĕ�(sh��)�����w(DMF) ƽ�_��ʹ�ö���ϵ�y(t��ng)ģ�K�܉��M(j��n)���Ԅ�NGS���Ʒ�Ƃ�ϵ�y(t��ng)��ͨ�^һ�N����ë��(x��)�ܽӿ������Ԍ�(sh��)�F(xi��n)Һ�w��DMF�O(sh��)���c�ⲿ���wģ�K�g�ĸ��؏�(f��)���D(zhu��n)�ƣ�ʹ����·�B�m(x��)����С�Θ�Ʒ�܉���һ��(g��)������ϵ�y(t��ng)��(n��i)�õ�̎�����@����ԓ���g(sh��)��(qi��ng)�{(di��o)��DMFԪ��ƽ�_��NGS ��Ʒ�Ƃ��������Ԅ��\(y��n)��ë��(x��)�ܽӿڵ�Ч������ë��(x��)�ܽӿ��cһ�NǶ��ʽ�ǽ��|늌�(d��o)�z�y���B�ӣ�DMF�O(sh��)�䌍(sh��)�F(xi��n)��Ŀ��(bi��o)������Ę�Ʒ����СҺ�ε��]�h(hu��n)�Ԅ�Ƭ�βɼ���NGS ���؏�(f��)�Δ�(sh��)���ľ��_Һ���Q�c��Ʒ��ϴͨ�^ʹ��һ�N�����Q����(sh��)�F(xi��n)��DMFƽ�_��ƽ��DNA�����ʞ�(80��4.8)%��
�������ǻ���ȱ�������ڙC(j��)�wȱ���ǻ�����������ҪӰ푵�N ���P(gu��n);����ɵ�������40% ���������ǻ���ȱ�ݲ������\���^���Л]�еõ�����ˮƽ�Ĵ_�J(r��n)��Jones ��ͨ�^�ѽ�(j��ng)��(d��o)���������ǻ���ȱ�ݻ������һ���y���(y��n)�C���о����ķ���ˮƽ������\���������ǻ���ȱ�ݵ�ˮƽ��12��δ֪�ӱ��²�ͻ׃���������ǻ���ȱ�ݲ���������Ԍ����M(j��n)��NGS�(y��n)�C���քe����RainDance �cFluidigm ƽ�_( dPCR) �M(j��n)�����и�����Ŀ��(bi��o)�U(ku��)����SOLiDƽ�_���ڜy��͔U(ku��)���a(ch��n)�����M(j��n)��ͨ�^NextGENe �M(j��n)��������Ϣ�W(xu��)�������Y(ji��)��������12��(g��)��Ԍ��յ��²�ͻ׃ͨ�^NGS�����Դ_�J(r��n)���ڲ����\���^������NGS�İl(f��)չʹ��(sh��)�(y��n)���\�������c��������(g��)������ȳɱ����ĸ������ٶȸ�����Ч�ʸ������R����(sh��)�(y��n)�ҵ���һ���y��(sh��)��(j��)�����Y(ji��)��ͬ��֧���@�(xi��ng)���g(sh��)���ƏVʹ���@�(xi��ng)���g(sh��)���P(gu��n)��Ҫ��
4 ���Y(ji��)�cչ��
����W(xu��)�Ļ��A(ch��)�о��ͷ��Ӽ��g(sh��)��ǰ�M(j��n)���S�������_���`���Ĝy�����g(sh��)�l(f��)չ��ֵ��һ����ǣ�dPCR���Мy����(d��)�����c�o���κ�У��(zh��n)������c(di��n)����ˣ�ԓ���g(sh��)�ǝ��ڵĺ���y������(zh��n)����������ԭ���Ϟ����Ӌ(j��)���ṩ�˱��C���������������dPCR����^�����������ܜ�(zh��n)�_����Ŀ��(bi��o)DNA���ṩ�ɿ��Ķ�����(sh��)��(j��)���̘I(y��)��dPCR �x��( ��Fluidigm ��˾��BioMark System)�Ĵ������F(xi��n)�M(j��n)һ���ƄӺ͔U(ku��)����ԓ���g(sh��)�İl(f��)չ�͑�(y��ng)�÷�����
dPCR���g(sh��)���䑪(y��ng)���@�ˆη��Ӷ������g(sh��)�ĝ��������������wdPCR�͕�ͻ�Ʒ���(y��ng)�ٶȺ��w�e�����ƣ���(sh��)�F(xi��n)�Ԅӻ���ͨ���đ�(y��ng)��������y��?q��)��ǻ���dPCR�Ćη��ӔU(ku��)�����g(sh��)����Ҫ�đ�(y��ng)���I(l��ng)����dPCR�Ŀ�¡�U(ku��)�����Ԝp����һ���y��ĕr(sh��)�g�ͳɱ�����ʹ��(g��)�˻���M�y����Ԍ�(sh��)�F(xi��n)���҂������ڲ��h(yu��n)�Č������@�(xi��ng)���g(sh��)�İl(f��)չ�����η��Ӻ���U(ku��)���I(l��ng)��a(ch��n)�����h(yu��n)Ӱ����ڷ�������W(xu��)���t(y��)�W(xu��)�Ȼ��A(ch��)�о��͑�(y��ng)�÷���l(f��)�]�����������
�����īI(xi��n)
[1] Keer J T , Birch L. Essentials of Nucleic Acid Analysis: A Robust Approach. London: Royal Society of Chemistry, 2008
[2] Zhang C, Xing D. Single-molecule DNA amplification and analysis using microfluidics.Chem Rev, 2010, 110 (8): 4910-4947
[3] Vogelstein B, Kinzler K W. Digital PCR. Proc Natl Acad Sci USA,1999,96(16): 9236-9241
[4] Bhat S, Herrmann J, Armishaw P, et al. Single molecule detection in nanofluidic digital array enables accurate measurement of DNA copy number. Anal Bioanal Chem, 2009, 394(2): 457-467
[5] Bustin S A, Benes V, Garson J A, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem , 2009, 55 (4 ): 611-622
[6] Burns M, Burrell A, Foy C. The applicability of digital PCR for the assessment of detection limits in GMO analysis. European Food Research and Technology, 2010, 231 (3 ): 353-362
[7] Sykes P J , Neoh S H , Brisco M J, et al. Quantitation of targets for PCR by use of limiting dilution. Biotechniques, 1992,13(3): 444
[8] Olga K, Irina L, James B, et al. Nanoliter scale PCR with TaqMan detection. Nucleic Acids Res, 1997,25 (10 ): 1999-2004
[9] Devin D, Hai Y, Giovanni T, et al. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations . Proc Natl Acad Sci USA, 2003,100 (15): 8817-8822
[10] Diehl F, Li M, Dressman D, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci USA, 2005, 102 (45): 16368-16373
[11] Frank D, Meng L, Yiping H, et al. BEA Ming: single-molecule PCR on micro particles in water-in-oil emulsions. Nat Methods, 2006, 3 (7): 551-559
[12] Liu J , Hansen C, Quake S R. Solving the " world-to-chip" in terface problem with a microfluidic matrix. Anal Chem,2003, 75(18): 4718-4723
[13] Dube S, Qin J , Ramakrishnan R. Mathematic alanalysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLoS One , 2008, 3 (8): e2876
[14] Heyries K A, Carolina T, Michael C, et al. Megapixel digital PCR. Nat Methods, 2011,8 (8): 649-651
[15] Sanders R, Jim F, Huggett, et al. Evaluation of digital PCR for absolute DNA quantification. Anal Chem, 2011, 83(17): 6474-6484
[16] Heid C A, Stevens J, Livak K L, e t al. Real time quantitative PCR. Genome Res, 1996, 6 (10): 986-994
[17] Morrison T B, Weis J J, Wittwer C T. Quantification of low-copy transcripts by continuous SYBR Green I monitoring during amplification. Biotechniques, 1998, 24(6): 954-958 , 960 , 962
[18] �w�\�s, �����, ���ٴ�, ��.����MGB̽���ɳ����ԭ�w��(sh��)�r(sh��)PCR�z�y�еđ�(y��ng)��. ���ﻯ�W(xu��)�c���������M(j��n)չ, 2003, 30(3): 466�C 470 Zhao J R, Bai Y J , Wang S C, et al. Prog Biochem Biophys, 2003,30(3): 466-470
[19] Corbisier P, Somanath B, Lina P, et al. Absolute quantification of genetically modified MO N810 maize(Zea mays L.) by digital polymerase chain reaction.Anal Bioanal Chem, 2009, 396 (6):2143-2150
[20] Terry C F, Shanahan D J , Ballam L D, et al. Real-time detection of genetically modified soya using Lightcycler and ABI 7700 platforms with TaqMan , Scorpion, and SYBR Green�� chemistries. J AOAC international, 2002, 85 (4): 938- 944
[21] Pohl G, Shih I M. Principle and applications of digital PCR. Expert Re v Mo l Di agn , 2 00 4, 4(1 ): 41- 47
[22] Oehler V G, Qin J, Ramakrishnan R, et al. Absolute quantitative detection of ABL tyrosine kinase domain point mutations in chronic myeloid leukemia using a novel nanofluidic platform and mutation-specific PCR. Leukemia, 2008, 23(2): 396-399
[23] Chan M, Mei W C, Ting W L, et al . Evaluation of nanofluidics technology for high-through put SNP genotyping in a clinical setting. J Mol Diagn, 2011, 13 (3): 305-312
[24] Ropers H H. New perspectives for the e lucidation of genetic disorders . Am J Hum Genet,2007, 81(2 ): 199-207
[25] Lupski J R. Genomic rearrangements and sporadic disease. Nat
Genet, 2007, 39: S43-S47
[26] Slamon D J, Clark G M , Wong S G, et al . Human breast cancer:correlation of relapse and survival with amplification of the HER-2 /neu oncogene .Science, 1987, 235 (4785): 177
[27] Qin J, Jones R C, Ramakrishnan R. Studying copy number variations using a nanofluidic platform. Nucleic Acids Res, 2008,36 (18): e116
[28] Lo Y M D. Noninvasive prenatal detection of fetal chromosomal aneuploidies by maternal plasman ucleic acid analysis: a review of the current state of the art. BJOG: An International J Obstetrics&Gynaecology, 2009, 116 (2): 152-157
[29] Lo Y M D, Fiona M F, Chan K C A, et al. Digital PCR for the molecular detection of fetal chromosomal aneuploidy. Proc Natl Acad Sci USA, 2007, 104(32): 13116 -13121
[30] Fan H C, Quake S R. Detection of aneuploidy with digital polymerase chain reaction. Anal Chem, 2007, 79 (19): 7576-7579
[31] Lo Y M, Nancy B Y T , Rossa W K C, et al. Plasma placental RNA allelic ratio permits noninvasive prenatal chromosomal aneuploidy detection. Nat Med , 2007, 13 (2): 218-223
[32] Lun F M, Allen C K, Yeung L T, et al. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem, 2008, 54(10): 1664-1672
[33] ����?zh��n)?��ԣ��.���ϾC�����ğo��(chu��ng)�a(ch��n)ǰ�\���о��M(j��n)չ. �Ї���(y��u)���c�z���s־, 2010,18(4): 137-139
Guo Q W, Zhou Y L. Chin J Birth Health & Hered, 2010, 18(4):137-139
[34] BioMark. BioMark Advanced Development Protocol 10. Absolute quantitation using the digital array; Fluidigm (Fluidigm Corporation, S.F)
[35] White A K, Michael V,Oleh I P, et al. High-through put micro fluidic single-cell RT-qPCR. Proc Natl Acad Sci USA, 2011,108(34): 13999-14004
[36] Spurgeon S L, Jones R C, Ramakrishnan R. High throughput gene expression measurement with real time PCR in a microfluidic dynamic array. PLoS One , 2008, 3(2): e1662
[37]Warren L,David B, Irving L W ,et al.Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc Natl Acad Sci USA , 2006,103(47):17807-1 7812
[38] Guo G , Huss M, Tong G Q, et al. Resolution of cell fated ecisions revealed by single-cell gene expression an alysis from zygote to blastocyst. Dev Cell , 2011,18 (4): 675-685
[39] Ottesen E A , Jong W H, Stephen R Q, et al. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria.Science ,2006,314 (5804): 1464-1467
[40] Tadmor A D, Elizabeth A O , Jared R L, et al. Probing in dividual environmental bacteria for viruses by using microfluidic digital PCR. Science, 2011,333(6038): 58-62
[41] Braslavsky I, Hebert B, Kartalov E, et al. Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci USA,2003, 100 (7): 3960-39 64
[42] Eid J, Fehr A, Gray J, et al. Real-time DN A sequencing from single polymerase molecules . Science, 2009, 323 (5910): 133-138
[43] Harris T D, Buzby P R, Babcock H, et al. Single-molecule DNA sequencing of a viral genome. Science, 2008, 320(5872 ): 106 -109
[44] White R A , Blainey P C, Fan H C, et al . Digital PCR provides sensitive and absolute calibration for high throughput sequencing.BMC Genomics, 2009, 10:116
[45] Zernant J , Schubert C, Im K, et al. Analysis of the ABCA4 gene by next-generation sequencing . Invest Ophthalmol Vis Sci, 2011,52(11): 8479-8487
[46] Kim H , Bartsch M S, Renzi R F, et al. Automated digital microfluidic sample preparation for next-generation DNA sequencing. J Lab Autom , 2011, 16(6): 405-414
[47] Jones M A, Bhide S, Chin E, et al. Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation. Genet Med, 2011,13 (11): 921-932

- ���әz�y���g(sh��)�ڱ���ƌW(xu��)�I(l��ng)��đ�(y��ng)��ǰ��Ԕ��
- ��(sh��)�r(sh��)�ɹⶨ��PCRԭ���͑�(y��ng)��
- PCR��(bi��o)��(zh��n)��������Ԕ��(x��)��B
- �ɲľ��x���ٙz�y�x��ԭ������(y��ng)��
- FluoGene®ϵ�y(t��ng)����Ѫ�ͻ�������Ԅӻ�
- �P�ſ��ٶ���PCRϵ�y(t��ng)�ڿ���30min�y��ֲ���D(zhu��n)����z�y�đ�(y��ng)��
- �½�PCR��(sh��)�(y��n)������ăx������(sh��)�(y��n)���|(zh��)�������wϵ���O(sh��)
- �P�Ŷ���PCR�ڷN�|(zh��)�YԴ����M�W(xu��)�о��đ�(y��ng)��
- �غ�Ƽ��y�ɹⶨ��PCR�x�������Ϻ�Ľ�������չ
- ���հ��c����s��26���Ї����H���¼��g(sh��)�ɹ����ו�
- ������ҕ����¡PCR�x����ȫ��ˮ�a(ch��n)���ܸ�ِ����e�k
- չ����Ո��:�غ�Ƽ����������Ϻ�Ľ���չ��
- ��¡���k�����������t(y��)����е��(chu��ng)�°l(f��)չ����A�M��Ļ
- �غ�Ƽ������չ��ʮ���������i�I(y��)���[��
- �غ�Ƽ��y�����Ǯa(ch��n)Ʒ����2024�ϰ݇��H��(sh��)�(y��n)��չ
- ��¡�Ƽ���(sh��)�r(sh��)�ɹ�PCR�����xϲ������t(y��)����еע���C


Copyright(C) 1998-2024 �������ľW(w��ng) �Ԓ��021-64166852;13621656896 E-mail��info@bio-equip.com